

Journal of Creation 29(2):106–109, August 2015
Browse our latest digital issue Subscribe
Nylon-eating bacteria—part 3: current theory on how the modified genes arose
Since genes used by ‘nylon-eating bacteria’ surely arose subsequent to nylon manufacture, this has been argued as proof for evolution. In Part 3 of this series we interpret this phenomenon using Coded Information System Theory, emphasizing that change is not the same thing as random evolution. The nature and source of information-processing guidance producing useful change needs to be first understood to permit a rational discussion of the significance of biological novelty. We summarize here what is currently believed to lie behind modified amide-degrading genes and why the original frame-shift theory, refuted in Part 2 on the basis of data already available at the time it was published by Ohno, is no longer believed on the basis of new data.

In Part 11 and Part 22 of this series we discussed the phenomenon of degradation of waste products resulting from the production of nylon-6.3 Since the responsible enzymes E-I – E-III in all likelihood arose, or at least were fine-tuned, after the introduction of nylon-6, this was used to argue no creator is necessary, by claiming novel proteins could arise by chance.4,5,6
A characteristic of information is that intention can be expressed during later periods of time and in other locations. The divine creation worldview presupposes that forethought, and front-loaded information, whether encoded on DNA or otherwise,7 can explain the origin of novel features. Whether chance or design offers a better explanation for biological novelty should be easier to evaluate by understanding the mechanism by which it may have arisen and how it now works.
To illustrate, intron/exon splicing in eukaryotes generates a richer variety of valuable proteins than is possible for simpler prokaryotes. Chance or design? Only a tiny proportion of possible mRNA sequences are biologically useful, and correct intron/exon boundaries exist for hundreds of thousands of genes throughout nature. The potential for error is overwhelming. What could possibly drive the chance origin of such a scheme, which would be selected against again and again due to unsuitable outcomes? For the scheme to work, spliceosomes are needed ab initio, which are “composed of as many as 300 distinct proteins and five RNAs, making it among the most complex macromolecular machines”.8 Evolutionary scenarios must be plausible if they are to be taken seriously.
We saw in Part 2 that initially virtually all scientists accepted unquestioningly Ohno’s claim9 that the enzymes had been produced by reading frame-shifts from pre-existing genes. No critical thought was given to whether this possibility would be more consistent with chance or design. Dr Thwaites, a professor of biology at San Diego State University, wasted no time to gloat:
We’ve been trying to explain all this to the protein ‘experts’ at ICR for the last seven years. We have told them that new proteins could indeed form from the random ordering of amino
acids … . Now it has happened! Not one, but two, new proteins have been discovered. In all probability new proteins are forming by this process all the time … . DNA sequence suggests that a simple ‘frame-shift’ mutation could have brought about the chance formation of at least this one enzyme … . All of this demonstrates that Yockey, Hoyle and Wickramasinghe, the creationists, and others who should know better are dead wrong about the near-zero probability of new enzyme formation.10
In Part 2 we showed how the storyline and debates based around Ohno’s frame-shift theory and claim that genes arose from simple repetitive oligomers were formulated using evolutionary speculations masquerading as factual raw data.11 Since most of the literature on the Internet on the origin of E-I–E-III has not been removed or revised, anyone new to the subject is likely to believe the frame-shift mechanism is true.
In Part 2 we analyzed the reported data, already available at the time everyone accepted Ohno’s notion, and this led to serious doubt he could have been right. Since then, new research, which we will discuss here, has led to universal rejection of Ohno’s claim by the experts in the field. Incidentally, Dr Thwaites’ triumphant statement, above, is factually wrong, both in terms of the frame-shift theory and probability of random peptide sequences producing a useful protein.
Current belief about the origin of E-II
Aligning enzyme F-E-II (F for Flavobacterium) and F-E-II’ sequences was reported to display 46 amino acid alterations12,13 after inserting some putative indels. The variant also present, F-E-II’, had only 0.5% as much catalytic activity toward Ald (figure 1).
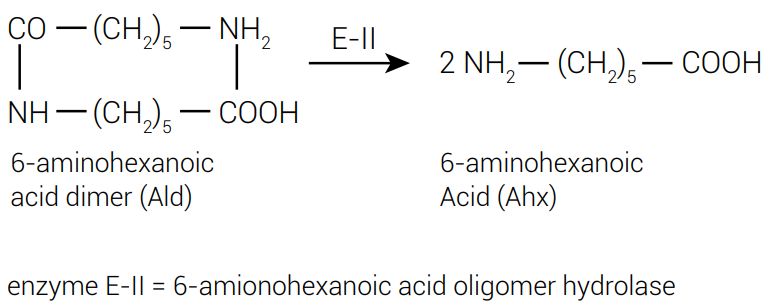
However, modifying only two amino acids in F-E-II’ to match those found in F-E-II (G181 to D, and H266 to N) was enough to increase the Ald-hydrolytic activity up to 85% of the level of the putative parental F-E-II enzyme.14 Alterations in the other 44 residues did not significantly increase the activity.
Negoro proposed that a common ancestral gene led to genes E-II and a variant E-II’, found on plasmid pOAD2 of Flavobacterium sp. K172.13 The authors were unable to produce good crystals of E-II’ for crystallographic studies, so they modified five amino acids in E-II’: T3A (i.e. Thr in position 3 was modified to Ala), P4R, T5S, S8Q, and D15G to match the residues found in E-II at these positions. The resulting gene, called Hyb-24, crystallized but displayed only the E-II’ level of activity. It is unlikely these five amino acid substitutions, all within fifteen residues from the end of the protein, would distort the folded structure of E-II’ significantly, so the crystallographic data should indeed reflect the folded structure of E-II’ also quite well.
A single alteration in Hyb-24, G181D, increased the Ald-hydrolytic activity15 by a factor of 11, and replacement of D (Aspartate) at position 181 in F-E-II with Asn, Glu, His, or Lys by site-directed mutagenesis drastically decreased the Ald-hydrolytic activity, but had little effect on the enzymatic activity on p-nitrophenylacetate (C2) and p-nitrophenylbutyrate (C4) esters.
The atomic coordinates of the deduced structure of Hyb-24, representing closely the protein product from E-II’, was used with the DALI server16 to find similar folded proteins stored in the Protein Data Bank.17 The two-domain structure was found to be similar to the folds of the penicillin-recognizing family of serine-reactive hydrolases (which have a characteristic Ser-X-X-Lys motif),12 especially to those of a d-alanyl-d-alanine-carboxypeptidase from Streptomyces (Z-score 26.7) and a carboxylesterase from Burkholderia (Z-score 24.1).18 Even though high Z-scores were found, the amino acid identity with Hyb-24 was very low, ranging from 10 to 19%.19
The E-II and Hyb-24 enzymes possessed hydrolytic activity toward C2 and C4 esters, and based on the spatial location and role of amino acid residues constituting the active sites for hydrolysis, the authors concluded that the ancestral gene was an esterase with a β-lactamase fold in which several amino acids were replaced in the catalyic cleft.20 Subsequent, more detailed, crystallographic analysis on Hyb-24 (PDB ID code: 1WYB) clarified the mode of catalysis down to the individual atom level and confirmed the mechanistic similarity with carboxylesterases. As further evidence, single substitutions of the key amino acids involved by site-directed mutagenesis led to the expected decrease in both nylon-oligomer and esterolytic hydrolysis, consistent with the three-dimensional model.21
The step-wise degradation process by the E-II enzyme (shown in figure 2) is very sensitive to structural details at the N-terminal part of the substrate.22 Inappropriate binding in this region prevented it from degrading a large number of amides having various N-terminal residues. However, the enzyme was far less specific for various carboxyl esters, and weak hydrophobic interactions implied by the crystallographic data may contribute to the substrate binding.23
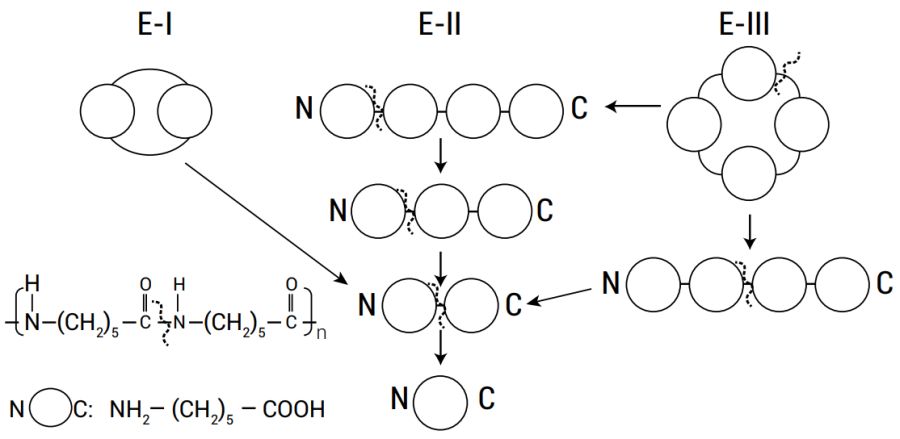
The data revealed convincingly how new activity towards nylon oligomer could easily arise with minimal changes while still retaining esterolytic functions.24 β-lactamases can be structurally very different but the S-X-X-K motif is apparently necessary.
What remains unknown are the exact source of the precursor gene and the mutations which had occurred, but Negoro reiterated how changing only two residues in E-II’ to match the amino acids found in E-II (Gly181 to Asp and His266 to Asn) increased the reactivity about 200-fold, to the level of E-II.25 In addition, the other 44 amino acid substitutions played no role in the enzyme activity.26 The same Shine-Dalgarno sequence was found in E-II and E-II’, and the same position for the initiation and termination codons. This suggests the two shared a common ancestor, and, presumably, E-II could have easily arisen with just a few mutations from a suitable precursor.
In Part 1 of this series we mentioned that E-III genes were found in several kinds of bacteria. These were labelled NylCp2 (from Arthrobacter), NylCA (from Agromyces) and NylCK (from Kocuria).27 The polypeptides encoded have 355 residues in all three cases, the same ATG initiation codon, and the same Shine-Dalgarno sequences (GGAGG). NylCA and NylCK have 5 and 15 amino acid substitutions, relative to the NylCp2 355 residue sequence, but their genes are believed to reside on the chromosome and not plasmids.28,29,30 These three enzymes are generated after a post-translational cleavage, which is a specific feature of the N-terminal nucleophile (N-tn) hydrolase family.
Four natural proteins very similar to NylA were found31 using the DALI program. Crystallographic analysis and comparison with the enzymatic mechanism of other N-tn hydrolases suggested the key step for NylC relies on just four residues,32 suitably held in place by the surrounding folded structure.
In Pseudomonas sp. NK87 the genes essential for the degradation of Acd (figure 3) were present on different plasmids. This implied that microorganisms were able to construct a sequential metabolic pathway of a synthetic xenobiotic compound by acquiring several plasmids encoding different enzymes. Later, these genes could be assembled into a single discrete unit through genetic rearrangements such as recombination and transposition.33,34 Assuming this interpretation is correct, the whole process occurred in a suspiciously rapid manner, implying some kind of informational guidance.

The P-E-I (Pseudomonas) and F-E-I (Flavobacterium) genes, which degrade Acd, were 99% identical at the DNA level. The history of these genes has not been reported, and plasmids are perhaps transporting the mutated genes back and forth rapidly, providing selective opportunities. In strain K172 the two genes are located on the same plasmid, but in strain NK87 on separate plasmids. This suggests plasmid distribution of genes is important to permit degradation of synthetic substances.
The E-II gene of Pseudomonas sp. NK87 (P-E-II) and Flavobacterium sp. K172 (F-E-II) were found to have 53% sequence similarity and 35% amino acid sequence similarity (after inserting several putative gaps to optimize the alignment).5 Another significant difference is that the codons used by the two enzyme variants for a key serine position was AGC in one case and TCG in the other, a difference of three single nucleotide mutations.35,36 Unfortunately, the authors did not speculate about what the ancestral common gene may have been.
No similarity was found between the F-E-I and F-E-II enzymes, or between the F-E-I and P-E-II enzymes. This implied the E-I and E-II enzymes arose independently,37 and not via a gene duplication or a frame-shift mutation.
Negoro admits to not knowing the molecular basis of nylon oligomer metabolism in strain PAO5502, but suggests that environmental stress favours this adaptation. He also provided a reference whereby micro-organisms may possess a cryptic gene which could be activated38 (and perhaps optimized through a few mutations). Bacterial enzymes which would be improved by a single mutation will inevitably occur. The plasmid pOAD2 has 45,519 bp, and if the mutation rate would be about 1 mutation per 1010 base pairs, typical of bacteria, then large populations of plasmid could already contain most acceptable single mutations, and there would also be many multiple mutational variants. Unlike chromosomal genes, plasmids need not necessarily be a permanent part of the bacteria, and damaged plasmids can be replaced by importing new ones or by carrying multiple copies. The tolerable mutational rate is likely to be higher than for chromosomal DNA.
Discussion
High mutation rates under stress
Micro-organisms were designed to be adaptable over a wide range of habitats and conditions. Adaptability includes fine-tuning by modifications of their DNA sequences. In the experiments described, various bacterial strains were subject to harsh conditions where survival would only be possible if a waste product from nylon production could be metabolized. Physiological details such as the availability of a given carbon source or other stress factors are known to affect mutation rate in bacteria, and so is the presence of transposable elements39 (such as the IS 6100 elements mentioned in Part 1), which can produce programmed recombinations.40,41
Mutational hotspots are regions on chromosomes where mutations occur rapidly, and the mutability of a single base can vary by more than 10,000-fold.42 Mutations need not be limited to reproduction, but also occurs in non-dividing cells, leading to the phenomenon of adaptive mutation, including the SOS mutagenic response, which involves more than SOS genes, associated with the protection, repair, replication, mutagenesis, and metabolism of DNA.43
Stress-induced mutagenesis can increase the mutation rate by several orders of magnitude under starvation conditions,44 and is clearly a regulated phenomenon. The main cause is the use of error-prone DNA polymerases V (umuCD) and IV (dinB), which transiently increase the rate of mutation.40 There are other forms for programmed DNA rearrangements, such as phase variation which is characterized by switching between high and low levels of activation of an ensemble of genes, which is also a tightly regulated process.40
Many bacterial populations include some members with a mutator phenotype. These bacteria can have a 10- to 50-fold increased mutation rate of up to 10,000 times, usually due to a defective methyl-directed mismatch repair system.36 These variants tend to be less fit and the same stress conditions which trigger their generation seem to also initiate programmed cell death.45 But nevertheless, under crisis conditions this subset could provide the means of generating a surviving new population.
Conclusions
Negoro and others provided compelling evidence the enzymes involved in degrading the synthetic materials created during manufacture of nylon-6 were derived easily from existing precursors. The detailed steps were not identified, but it seems likely that the reaction being catalyzed is not very challenging, as Behe already pointed out,46 and could be attained from many starting points and by many variants. The harsh nutritional challenges likely invoked pre-existing survival responses, which then led to increased mutations to survive at all costs.
Bacteria need to degrade and recycle organic substances into useful raw materials for themselves and other organisms. The huge population sizes, short generation times, robustness to a reasonable amount of mutations, and resultant ability to fine-tune general-purpose families of enzymes are part of the ecological design. If evolutionary principles really did lead to the vast complexity found in organisms, the examples produced as evidence must deal with the creation of complex proteins, molecular machines, organs and organisms, not merely the modification of existing proteins to utilize new sources of nutrients.
References and notes
- Truman, R., Nylon-eating bacteria—part 1: discovery and significance, J. Creation 29(1):95–102, 2015. Return to text.
- Truman, R., Nylon-eating bacteria—part 2: refuting Ohno’s frame-shift theory, J. Creation 29(2):78–85, 2015. Return to text.
- Kinoshita, S., Kageyama, S., Iba, K., Yamada, Y. and Okada, H., Utilization of a Cyclic Dieter and Linear Oligomers of ͼ-Aminocaproic Acid by Achromobacter guttatus KI 72, Agr. Biol. Chem. 39(6):1219–1223, 1975. Return to text.
- Thomas, D., New Mexicans for Science and Reason presents Evolution and Information: The Nylon Bug, www.nmsr.org/nylon.htm. Return to text.
- Musgrave, I., Nylonase Enzymes, The TalkOrigins Archive, ww.talkorigins.org/origins/postmonth/apr04.html, 20 April 2004. Return to text.
- Thwaites, W.M, New Proteins Without God’s Help, NCSE 5(2):1–3, 1985; ncse.com/cej/5/2/new-proteins-without-gods-help. Return to text.
- Truman, R., Information Theory—Part 3. Introduction to Coded Information Systems, J. Creation 26(3):115–119, 2012; creation.com/cis-3. Return to text.
- Nilsen, T.W., The spliceosome: the most complex macromolecular machine in the cell? BioEssays 25(12):1147–1149, 2003; onlinelibrary.wiley.com/doi/10.1002/bies.10394/pdf. Return to text.
- Ohno, S., Birth of a unique enzyme from an alternative reading frame of the pre-existed, internally repetitious coding sequence, Proc. Natl. Acad. Sci. USA 81:2421–2425, 1984. Return to text.
- Thwaites, ref. 6, p. 1. Return to text.
- “Since we know dinosaurs died out 65 million years ago, it is clear that this fossil could not …” The statement in quotation marks would be an example of an evolutionary assumption, not a fact. Return to text.
- Negoro, S. et al., Nylon-oligomer Degrading Enzyme/Substrate Complex: Catalyic Mechanism of 6-Aminohexanoate-dimer Hydrolase, J. Mol. Biol. 370:142–156, 2007. Return to text.
- Negoro, S., Ohki, T., Shibata, N., Mizuno, N., Wakitani, Y., Tsurukame, J., Matsumoto, K., Kawamoto, I., Takeo, M. and Higuchi, Y., X-ray crystallographic analysis of 6-aminohexanoate-dimer hydrolase: molecular basis for the birth of a nylon oligomer-degradation enzyme, J. Biol. Chem. 280:39644–39652, 2005. Return to text.
- Negoro et al., ref. 12, p. 144. Return to text.
- Negoro et al., ref. 13, p. 39651. Return to text.
- ekhidna.biocenter.helsinki.fi/dali_server. Return to text.
- www.rcsb.org/pdb/home/home.do. Return to text.
- Negoro et al., ref. 13, p. 39649, table 1. The protein identifiers in the Protein Data Bank are also provided there. Return to text.
- Negoro et al., ref. 13, p. 39647. Return to text.
- Negoro et al., ref. 13, p. 39644. Return to text.
- Negoro et al., ref. 12, p. 147. Return to text.
- Negoro et al., ref. 12, p. 153. Return to text.
- Negoro et al., ref. 12, p. 149. Return to text.
- Negoro et al., ref. 12, p. 154. Return to text.
- Negoro, S., Biodegradation of nylon oligomers, Appl. Microbiol. Biotechnol. 54:461–466, 2000. Return to text.
- Negoro, ref. 25, p. 464. Return to text.
- Negoro, A., Shibata, N., Tanaka, Y., Yasuhira, K., Shibata, H., Hashimoto, H., Lee, Y-H., Oshima, S., Santa, R., Oshima, S., Mochiji, K., Goto, Y., Ikegami, T., Nagai, K., Kato, D., Takeo, M. and Higuchi., Y., Three-dimensional Structure of Nylon Hydrolase and Mechanism of Nylon-6-Hydrolysis, J. Biol. Chem. 287:5079–5090, 2012. Return to text.
- Yasuhira, K., Tanaka, Y., Shibata, H., Kawashima, Y., Ohara, A., Kato, D., Takeo, M. and Negoro, S., 6-Aminohexanoate Oligomer Hydrolases from the Alkalophilic Bacteria Agromyces sp. Strain KY5R and Kocuria sp. Strain KY2, Appl. and Environ. Microbiol.73(21):7099–7102, 2007. Return to text.
- Yasuhira et al., ref. 28, p. 7100. Return to text.
- Negoro et al., ref. 27, p. 5080. Return to text.
- Negoro et al., ref. 27, p. 5083. Return to text.
- Negoro et al., ref. 27, p. 5084. Return to text.
- Kanagawa, K., Negoro, S., Takada, N. and Okada, H., Plasmid dependence of Pseudomonas sp. strain NK87 enzymes that degrade 6-aminohexanoate-cyclic dimer, J. Bacter. 171:3181–3186, 1989. Return to text.
- Kanagawa et al., ref. 33, p. 3185. Return to text.
- Kanagawa, K., Oishi, M., Negoro, S., Urabe, I. and Okada, H., Characterization of the 6-aminohexanoate-dimer hydrolase from Pseudomonas sp. NK87, J. Gen. Microbiol. 139:787–795, 1993. Return to text.
- Kanagawa et al., ref. 35, p. 793. Return to text.
- Kanagawa et al., ref. 35, p. 794. Return to text.
- Negoro, ref. 25, p. 465. Return to text.
- Martinez, J.L. and Baquero, F., Mutation Frequencies and Antibiotic Resistance, Antimicrob. Agents Chemother. 44(7):1771–1777, 2000. Return to text.
- Martinez and Baquero, ref. 39, p. 1774. Return to text.
- Kidwell, M.G. and Lisch, D.R., Transposable elements and host genome evolution, Trends Ecol. Evol. 15:95–99, 2000. Return to text.
- Drake, J.W., A constant rate of spontaneous mutation in DNA-based microbes, Proc. Natl. Acad. Sci. USA 88:7160–7164, 1991. Return to text.
- Janion, C., Inducible SOS Response System of DNA Repair and Mutagenesis in Escherichia coli, Int. J. Biol. Sci. 4:338–344, 2008. Return to text.
- Martinez and Baquero, ref. 39, p. 1773. Return to text.
- Martinez and Baquero, ref. 39, p. 1771. Return to text.
- An Interview with Dr. Michael J. Behe, www.ideacenter.org/contentmgr/showdetails.php/id/1449, accessed 18 May 2014. Return to text.









Readers’ comments
Comments are automatically closed 14 days after publication.