
Mutations are more harmful than we thought
Silencing the ‘wobble’ in the codon table
As regular readers of this website already know, there is a sea of change happening in evolutionary biology. As the complexity of life is being uncovered, the simplistic arguments from yesteryear are struggling to keep up. One of those tired old ideas is the thought that the genome is chock full of useless ‘junk’ DNA. We have written about the slow demise of junk DNA theory many times before, and how adherence to the concept has slowed scientific progress, but new revelations are continuing to pile up. In this case, it is the ‘wobble base’ that is taking a big hit. Let me explain.
As we all learn in biology class, there are many steps to making a protein. The cell starts with a protein-coding gene on the DNA and transcribes it into RNA. An amazing machine called a ribosome translates the RNA, three letters at a time, to make the protein. Those three-letter sets are called codons. The ribosome uses a set of adapter molecules called transfer RNAs (tRNAs). At the bottom of each tRNA is a set of three letters (the anti-codon) that complements one specific codon only. At the other end of each tRNA is an amino acid waiting to be added to the end of the growing protein. The ‘wobble base’ is the last letter in the codon. Since there are four nucleotides in DNA (A, C, G, and T), there are 64 (4 x 4 x 4) different codons. But there are only 20 amino acids (actually, 211) coded in our genes. This means that multiple codons can be used for the same amino acid (figure 1).
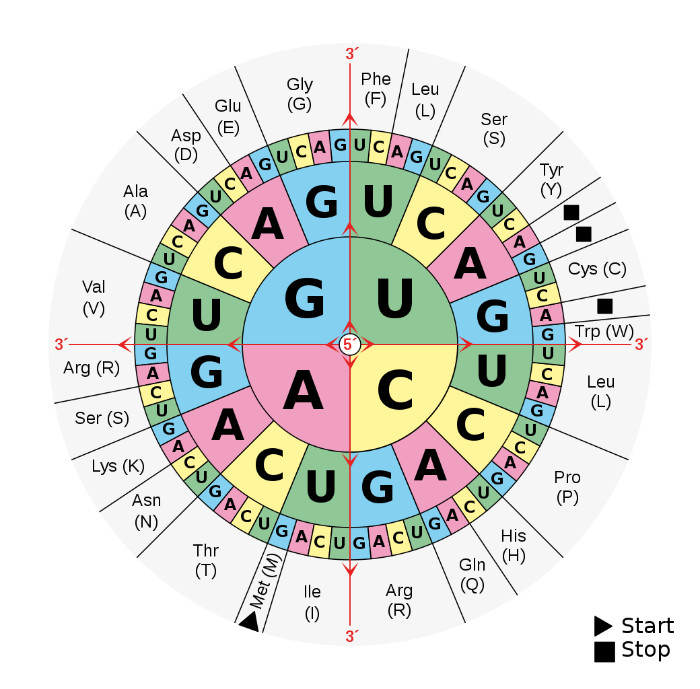
The codon table is also optimized to reduce the functional impact of a change from one amino acid to another. The system was set up so that when a mutation causes a switch to a different amino acid, the change is usually to an amino acid with a similar chemical state (e.g., similar charge, size, or polarity).2 God left little to chance here, so maybe it should not surprise us that the ‘wobble base’ is a bit more complicated than we first thought.
We have known since the 1960s that many mutations do not affect the amino acid sequence in proteins. The reason for this is that mutations will often cause a switch among codons for the same amino acid. For example, the DNA letter combinations GGA, GGC, GGG, and GGT all code for the amino acid glycine. You will notice that they all start with “GG” and that any of the four DNA nucleotides can be put in that last position. As I mentioned earlier, the last letter of each of these codons is called the ‘wobble base’. This is because, through mutation, the letter can wobble back and forth between any of the four DNA letters and the protein is not affected. Mutations at the wobble base are called ‘silent’ mutations. They are also called ‘synonymous’ mutations, as opposed to ‘non-synonymous’ mutations that change the amino acid sequence in the protein.
Scientific jargon can be confusing sometimes, but the words are often chosen for dramatic effect. Another term used when discussing the codon table is ‘degeneracy’.3 This makes it sound like evolution randomly stumbled upon an inefficient and mostly random way of coding for amino acids. In the past, it also made people think that most mutations did not matter. Thus, for many years, biologists thought that synonymous mutations were silent mutations and so were inconsequential. I suspect this slowed the search for function and hampered people’s ability to see the design in life. If the multiple codons used for certain amino acids are not ‘redundant’ but functional (e.g., controlling the rate of formation of a protein due to differing populations of tRNAs), this would inject another level of complexity into the optimisation of the genetic code. One’s choice of language can have important consequences: ‘silent’, ‘wobbly’, ‘synonymous’, ‘degenerate’, ‘redundant’—can you see a pattern? At every step evolutionists have sought to minimize the possibility that life was designed.
The paradigm collapses
Hints that they were wrong have been trickling in over the years. As early as 2007, we knew that changing the third codon letter in a few bacterial proteins caused the protein to misfold and become useless. It was thought that the switch to a rare codon caused a pause in protein translation as the cell searched for the correspondingly rare tRNA, which would then cause the protein to misfold while translation was paused.4,5
There are many other reasons why a synonymous mutation (i.e., a wobble) could be harmful. The sequence of letters in a protein-coding gene does more than just code for the amino acids. For example, any given letter could be a site for histone binding, an intron/exon splice site, or an attachment point for any number of gene regulators. I wrote about this in my 2010 article Splicing and dicing the human genome. Soon after this, scientists coined the term ‘duons’ to describe nucleotides that have multiple simultaneous functions.6 Creation Ministries International (CMI) speaker and scientist Philip Bell wrote about this in his review of Stephen Myer’s book Darwin’s Doubt, saying:
“The rub is that this code is written right on top of the existing DNA code! In other words, it now appears that many of the 3-letter codons have a dual function—so have been appropriately dubbed ‘duons’—another nail in the coffin of neo-Darwinian evolution and certainly something Meyer would have included in Darwin’s Doubt but for its recency.”
This was based upon a University of Washington press release, part of which states:
“The UW team discovered that some codons, which they called duons, can have two meanings, one related to protein sequence, and one related to gene control. These two meanings seem to have evolved, in the minds of the researchers, in concert with each other. The gene control instructions appear to help stabilize certain beneficial features of proteins and how they are made.
The discovery of duons has major implications for how scientists and physicians interpret a patient’s genome and will open new doors to the diagnosis and treatment of disease.”7
Getting back to the main subject, recent studies on the wobble bases brought in even more surprises. A 2011 review on our understanding of the subject concluded, “Synonymous mutations, once thought to be ‘silent’, are now increasingly acknowledged to be able to cause changes in protein expression, conformation and function.” They noted that silent mutations make a substantial contribution to human diseases and that up to 10% of human genes contained at least one region where synonymous mutations could potentially be harmful.8 This was an underestimate.
In an as-yet unpublished manuscript, another group studying silent mutations detected a strong correlation between the choice of codon and the resulting shape of the folded protein. They commented that there were “… vast biological implications should coding be shown to directly shape protein conformation.”9 If the ‘wobble base’ hypothesis were to fall, there are ‘vast biological implications’? Indeed so.
Another group of scientists compiled a list of 659,194 synonymous mutations that were found in human cancers. After assessing the functional impact of these mutations, they concluded, “Synonymous mutations have been viewed as silent mutations, since they only affect the DNA and mRNA, but not the amino acid sequence of the resulting protein. Nonetheless, recent studies suggest their significant impact on splicing, RNA stability, RNA folding, translation or co-translational protein folding.”10
The final nail in the coffin?
Consider the title of another new paper in Nature (June 2022) “Synonymous mutations in representative yeast genes are mostly strongly non-neutral.”11 It will take a little time to unpack the title of this paper. First, they were studying yeast. This does not sound impressive until you realize that yeasts have radically different genetics than bacteria. In fact, in many ways yeasts are genetically similar to humans and these single-celled organisms have often been used to model what might happen in more complex species.
Second, the phrase “mostly strongly” is an odd choice, unless you were trying to get a strong point across. They are saying that instead of being silent, the great majority of wobble base mutations have a strong effect on the organism. This is not supposed to be true! Evolutionists were OK with a few exceptions to their rule, but now they are OK with saying that most mutations that do not affect the choice of amino acid are bad?
As the CMI scientists were discussing this, Dr. Samuel Gan, a member of the Singapore Friends of CMI group who was interviewed in Creation magazine in 2021, wrote,
“The article essentially suggests that the wobble hypothesis needs a rethink and that there is much that we still need to do. It seems to me that this represents yet another barrier to cross-kind evolution, although we are in the early days yet.”
The implications are clear: if most silent changes are actually harmful to the organism, strong barriers to evolutionary change would be set up. ‘Wobbles’ in the third codon base have turned out to be important after all and appeals to ‘redundancy’ and ‘degeneracy’ were a big mistake! The evolutionary community is only just now starting to realize this, and some of them are not liking what they are seeing.
Finally, they use the phrase “non-neutral”. Neutralism was an evolutionary escape mechanism, coming on the heels of what is now called Haldane’s dilemma. In the 1950s, Haldane realized that natural selection could only do so much. He reasoned that selection could only explain a few hundred differences between humans and chimps, but we now know that there are tens of millions of genetic differences between us. In the late 1960s, neutral theory was developed, and even though it remained controversial, it was still the ruling paradigm for several decades. They reasoned that, since only about 2% of the genome codes for protein, the other 98% was free to mutate and drift at random. In this way, a few hundred selectable mutations in the protein-coding sections and an untold number of mutations in junk DNA might be able to explain the vast differences between the two species. With these things in mind, then, for these authors to claim that almost all silent mutations are strongly non-neutral is tantamount to throwing down a gauntlet. And they did this in Nature, the world’s most prestigious science journal.
They discovered that fully three-quarters of the more than 1000 synonymous mutations they studied resulted in a significant reduction in the fitness of their yeast strains. They also noticed that the average synonymous mutation had about the same (negative) effect as the average nonsynonymous mutation. They conclude their abstract with,
“The strong non-neutrality of most synonymous mutations, if it holds true for other genes and in other organisms, would require re-examination of numerous biological conclusions about mutation, selection, effective population size, divergence time and disease mechanisms that rely on the assumption that synonymous mutations are neutral” (emphasis added).
An editorial in that same issue of Nature was simply titled “Mutations matter even if proteins stay the same.”12
However, the conclusions of this paper have been strongly contradicted in an unpublished manuscript that appeared on the preprint server bioRxiv.13The authors claimed that the work of Shen et al. contradicts “well-established findings from a broad range of fields and approaches.” This in itself was not a problem, for many scientific advances contradict what was once thought to be true. However, they pointed out that the experiment may have lacked proper controls. Specifically, Shen et al. used the CRISPR-Cas9 system to edit the genomes of the yeasts but they did not control for the off-target effects that CRISPR is known to produce. Apparently, there was no way to gauge any unintended genetic changes because they only used one wild-type yeast strain for comparison and it had not gone through the same process as the experimental versions. They also did not sequence the entire genome of the edited strains.
One wonders how Shen et al.’s work could have made it into the world’s most prestigious scientific journal if it had such glaring errors. Yet, even if the study is eventually retracted, this does not mean that ‘wobble’ bases are neutral. Polyfunctionality is still a thing. Synonymous changes are still known to frequently cause changes in protein expression, conformation, and function. They are known to affect mRNA splicing, RNA stability, RNA folding, protein translation, and co-translational protein folding. While we would hope that more functions can be found for ‘wobble’ bases, we know enough to conclude that they are more important than once believed.
Neutral alleles?
We now understand that most changes in the protein coding region should be bad for the organism. This points toward genetic entropy, an idea which leverages the beliefs and language of the neutralist position to make a strong case against it. Using their own ideas, genetic entropy contradicts their position because, given the best-case scenario for evolution (i.e., most mutations are nearly neutral), their case still falls apart when critically examined. IF, however, many of those neutral mutations are not actually neutral, the case against evolution becomes insurmountable. Thus, on the one hand, we have already killed evolution on mathematical grounds (see A successful decade for Mendel’s Accountant). On the other hand, we can now drive more nails into the coffin of neo-Darwinism. We can seriously ask, “So, you need selectively neutral alleles? What neutral alleles?”
References and notes
- The 21st amino acid is selenocysteine. It has its own transfer RNA that uses the codon UGA. This was thought to always be a STOP codon, but multiple gene sequences contain this codon in the middle of the gene. There are many, many other amino acids that are found in our proteins, but these are added to the protein by chemically modifying the amino acids after the protein is manufactured. The selenocysteine tRNA is initially charged with the amino acid serine (using seryl-tRNA ligase) and is then modified to selenocysteine with an enzyme called selenocysteine synthase. Return to text.
- Truman, R. and Terborg, P., Genetic code optimisation: Part 1, J. Creation 21(2):90–100, 2007; creation.com/genetic-code-optimisation-1. Return to text.
- The term degenerate is also used in quantum mechanics. It simply means “having equal energy”, referring to more than one quantum state. In general, higher symmetry leads to more degeneracy. Lowering symmetry can result in splitting degenerate levels, e.g., by a magnetic field or a coordination complex. Similarly, degeneracy in genetics simply means “coding for the same amino acid”. It does not mean “decrepit”. Instead, it means “lacking distinctness”. Return to text.
- Kimchi-Sarfaty, C. et al., A “silent” polymorphism in the MDR1 gene changes substrate specificity, Science 315:525–528, 2007. Return to text.
- Tsai, C.-J. et al., Synonymous mutations and ribosome stalling can lead to altered folding pathways and distinct minima, J. Mol. Biol. 383(2):281–291, 2008. Return to text.
- Stergachis, A.B. et al., Exonic transcription factor binding directs codon choice and affects protein evolution, Science 342(6164):1367–1372, 2013. Return to text.
- Seiler, S., Scientists discover double meaning in genetic code, washington.edu, 12 Dec 2013. Return to text.
- Sauna, Z.E. and Kimchi-Sarfaty, C., Understanding the contribution of synonymous mutations to human disease, Nat. Rev. Genet. 12:683–691, 2011. Return to text.
- Rosenberg, A., Marx, A, and Bronstein, A., Codon-specific Ramachandran plots show amino acid backbone conformation depends on identity of the translated codon, doi.org/10.21203/rs.3.rs-1089201/v1, 2022. Return to text.
- Sharma, Y. et al., A pan-cancer analysis of synonymous mutations, Nat. Comm. 10(2569), 2011 | doi.org/10.1038/s41467-019-10489-2. Return to text.
- Shen, X. et al., Synonymous mutations in representative yeast genes are mostly strongly non-neutral, Nature 606:725–731, 2022. Return to text.
- Sharp, N., Mutations matter even if proteins stay the same, Nature 606:657–659, 2022. Return to text.
- Kruglyak, L. et al., No evidence that synonymous mutations in yeast genes are mostly deleterious, bioRxiv, 15 Jul 2022 | doi: 10.1101/2022.07.14.500130.. Return to text.



Readers’ comments
Comments are automatically closed 14 days after publication.